Research
Gene Regulation: Understanding the Principle, Engineering the System, and Evolving the Medicine.
Research
The mission of our laboratory is to elucidate the function and mechanism of genome and RNA and to apply them to better understand the molecular mechanisms of cancer and to develop the new approaches to cancer therapy. How do genes work? This fundamental question is crucial to understand the mechanisms of diverse diseases including cancer, which involve the interactions between genetic factors and environmental factors. Through an integrative omics analysis based on next-generation sequencing, bioinformatics, and genome/RNA engineering, we aim to get a deeper understanding of the relationships between genome, epigenome, transcription, and RNA to uncover the operating principles of gene regulation and disease mechanisms. We also aim to facilitate cancer precision medicine based on comprehensive omics analysis and develop new methodologies and technologies to advance this.
1.Super-enhancer and Phase Separation
How do different cell types with the same genome establish cell-type-specific transcriptional programs? Cell-type-specific transcriptional programs are activated by the cell-type-specific enhancers bound by master transcription factors. Super-enhancers are emerging subclass of distal enhancers which regulate cell identity and disease processes. We have focused on the function of super-enhancers from the stand point of RNA. By integratively analyzing the relationships between super-enhancers and miRNAs, we have reported that super-enhancers have central roles in driving the function of cell-type-specific and evolutionally conserved miRNAs and maintaining their regulatory networks (Suzuki et al, Cell, 2017). In addition, we reported that super-enhancers facilitate processing of miRNA precursors, revealing unrecognized higher-order property of super-enhancers.
Recently, liquid-liquid phase separation (LLPS) has been attracting attention as the working principle of intracellular non-membrane structures, and it has become clear that LLPS and intracellular structures formed by LLPS, i.e., biomolecular condensates, play important roles in various cellular processes such as gene regulation and signal transduction. Based on various studies on super-enhancers, a model has been proposed in which strong transcriptional activation is induced by the accumulation of transcription factors, Mediator, and other transcriptional regulators at the super-enhancer region via LLPS (Suzuki et al, Cancer Science, 2022). We are currently exploring the relationship between intracellular phase separation and genome and transcriptional regulation through comprehensive data analysis of intrinsically disordered regions, which are drivers of intracellular phase separation, and development of new technologies, which we will apply to disease research. We are also conducting research to analyze the function of super-enhancers using machine learning.

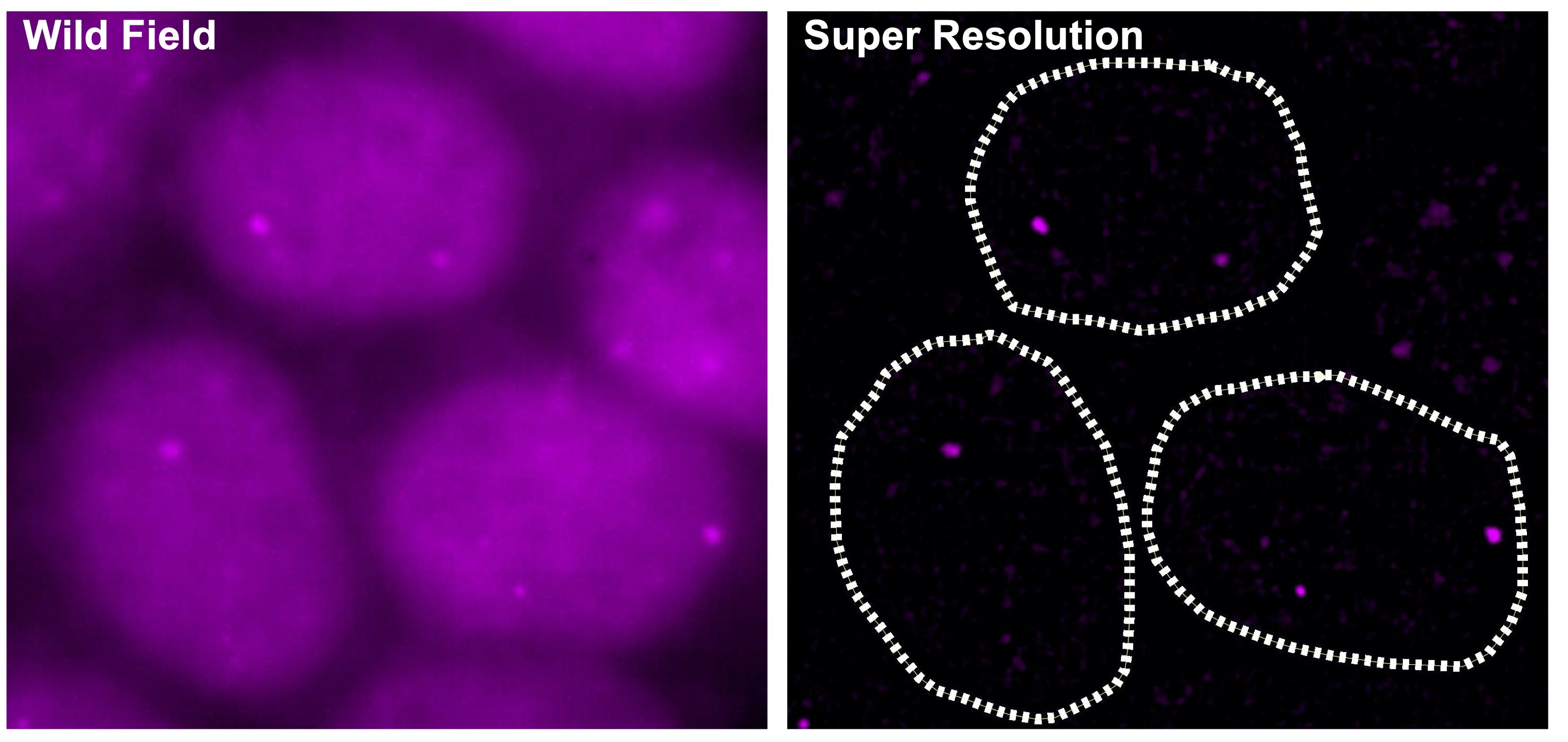
2.Targeting eccDNAs in cancer
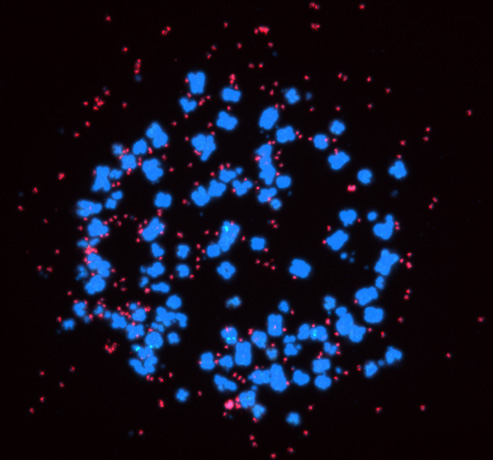
Super-enhancers also have a very important role in the transcriptional regulation of cancer cells. Based on a comparison of super-enhancers in normal cells/tissues and cancer cells, we have found that widespread alterations of neighbor SEs, including a loss of super-enhancers associated with tumor-suppressive miRNAs and a formation of super-enhancers associated with tumor-promoting miRNAs, were observed for multiple miRNA genes associated with cancer (Suzuki et al, Cell, 2017). In association with the identification of the first human disease caused by gain-of-function mutations in miRNAs (Grigelioniene et al, Nature Medicine, 2019), we also report new directions for big data analysis of functional genomics in cancer research (Suzuki et al. Nature Genetics, 2018). Inhibitors of BRD4 and CDK7, which are involved in transcriptional regulation by super-enhancers, are being considered for cancer therapy, but on the other hand, super-enhancers naturally function in normal cells as well, raising the issue of selectivity and specificity of such treatments. Recently, we have thus focused on extrachromosomal circular DNAs (eccDNAs) to develop more specific transcription-targeted therapies for cancer cells.
In cancer, chromosomal translocations, amplifications, and mutations contribute to aberrant activation of enhancers and super-enhancers, and the aforementioned super-enhancer-targeted therapies also assume enhancers within chromosomes as their targets. Following the description of extrachromosomal DNAs called double minutes (DM) since around 1980, the existence of extrachromosomal circular DNAs (eccDNAs) has recently been highlighted since around 2017. The presence of oncogenes and (super-)enhancers in eccDNAs, which is more transcriptionally active than chromosomal DNA and is not found in normal cells, may make it a potential target for more selective transcriptional targeting therapies. In addition, cases with eccDNAs have been reported to have a poorer prognosis, and eccDNAs been been proposed to contribute to cancer evolution, cancer cell heterogeneity, and the acquisition of drug resistance associated with heterogeneity. The struggle between cancer and therapy is a perennial issue in cancer treatment, and no cancer therapy has yet been developed that would target eccDNAs. We are currently developing more specific transcription-targeted therapies that target eccDNAs.
3.Understanding transcription cycle based on RNA diversity
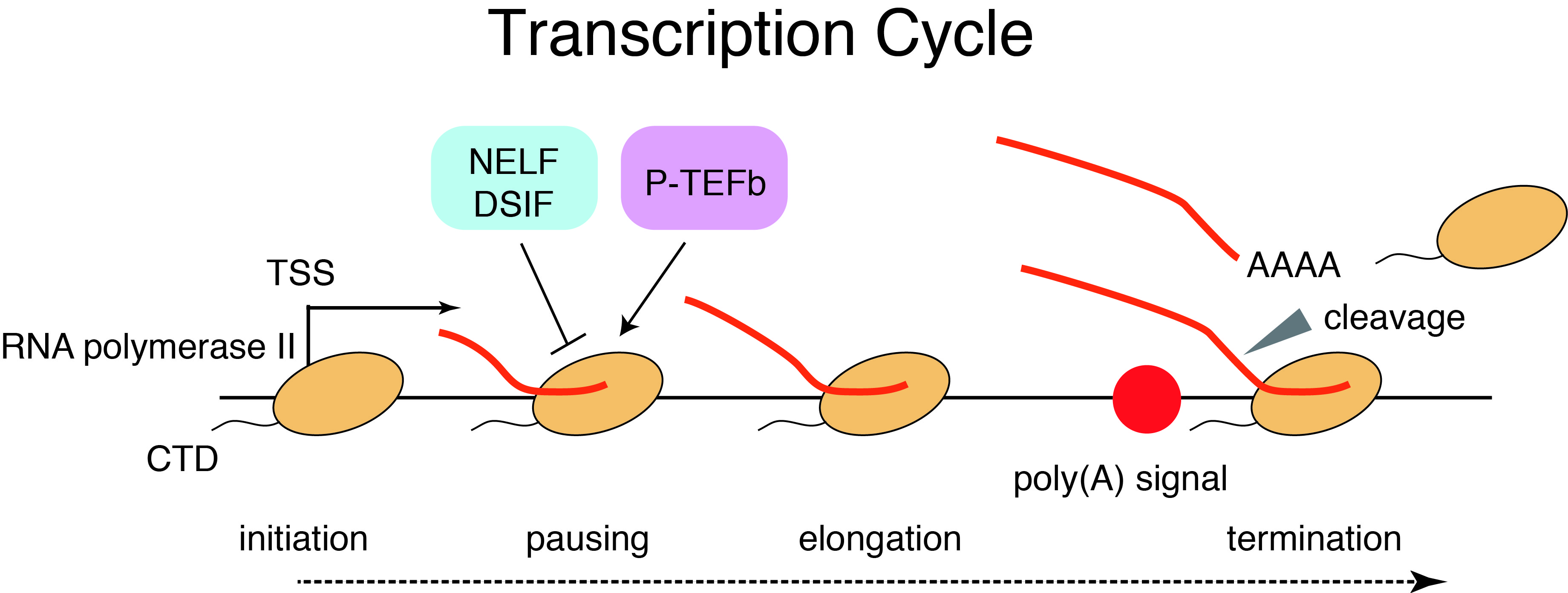
Toward better understanding of enhancer function, it is important to understand the dynamics of transcription across enhancers and the transcriptional impacts of enhancers on cognate promoters. Divergent transcription by RNA polymerase II occurs at many enhancers and promoters, yielding both coding mRNAs and diverse ncRNAs including eRNAs and uaRNAs. Furthermore, transcription by RNA polymerase II is regulated at multiple steps including initiation, pausing, pause release, elongation, and termination (“transcription cycle”). We have investigated the detailed mechanisms of transcription by integrating genome science and information science.
Protein-coding promoters are characterized by elongation of RNA polymerase II in the sense direction and early termination in the antisense direction. Such transcriptional directionality is modulated by asymmetrical distribution of U1 splicing motif and poly-adenylation signal (PAS motif) (U1-PAS axis). We have previously reported that premature PAS termination also frequently occurs in the sense direction and coincides with RNA polymerase II pause sites associated with +1 stable nucleosomes (Chiu et al, Mol Cell, 2018). Thus, promoter proximal Pol II pausing consists of two processes: transcription start site (TSS)-proximal pausing and +1 stable nucleosome pausing.
Recent advances in cancer genome studies have uncovered new class of cancer driver mutations affecting RNA processing and transcription. On the other hand, their functional significance has not been fully investigated in many cases. In myelodysplastic syndromes (MDS) and related neoplasms, the members of the cohesin complex including STAG2 are frequently mutated. In a recent collaboration study, we have importantly found that combined loss of STAG2 and master transcription factor RUNX1 synergistically attenuates enhancer–promoter loops, leading to development of MDS, and that cohesin deficiency preferentially downregulates genes with high basal transcriptional pausing, explaining the feature of selective gene dysregulation (Ochi et al, Cancer Discovery, 2020).
We are currently focusing on super-enhancers, RNA modifications, and transcription termination mechanisms to explore new mechanisms of transcriptional regulation and to apply them to cancer research.
4.Disease research using multi-omics analysis and single cell analysis

Our laboratory has promoted a wide range of research to explore the mechanisms of diseases through integrated analysis of large data sets using next-generation sequencers. In particular, we have many years of experience in the analysis of microRNAs, epigenetics, super-enhancers, and transcriptional regulation. Currently, by integrating single cell analysis in addition to these multilevel omics analyses, we are advancing the pathological analysis of not only cancer but also various other diseases.
5.MicroRNA and Cancer
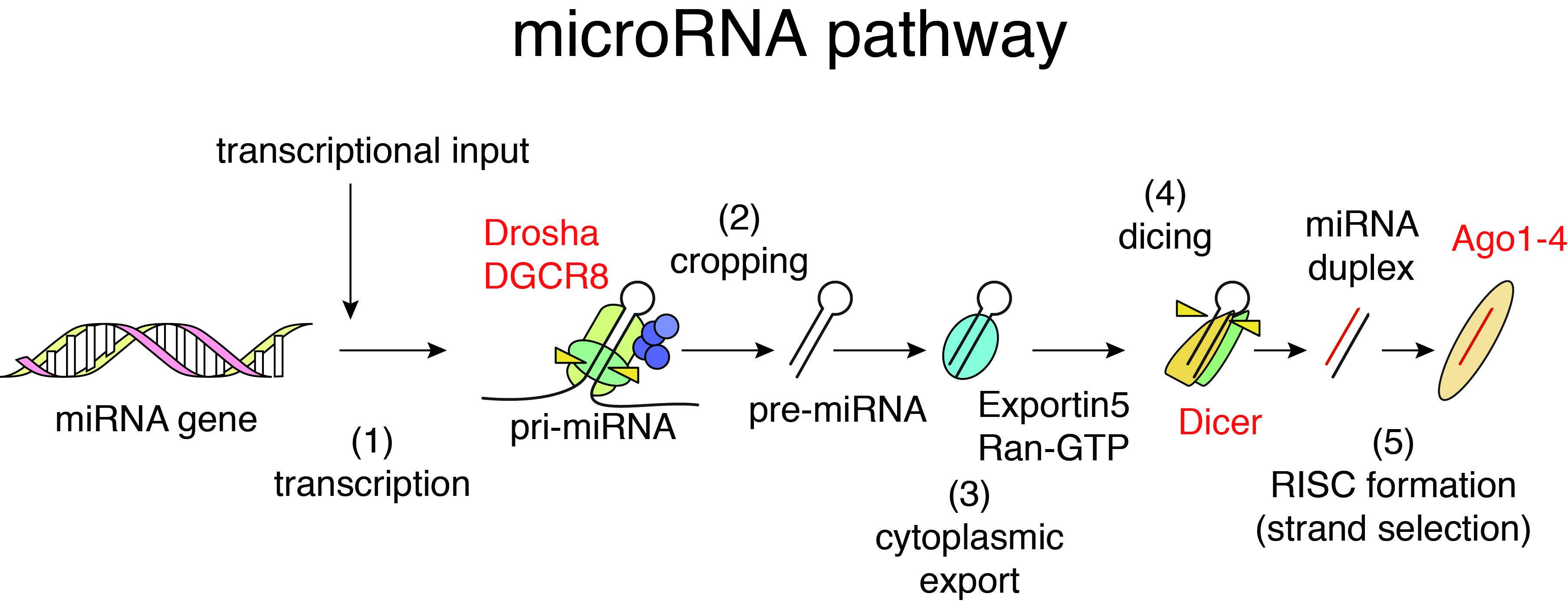
Function of many genes is governed by the flow of genetic information from DNA to RNA to protein, the central dogma of molecular biology. On the other hand, non-coding regions of the genome are pervasively transcribed and yield thousands of non-coding RNAs (ncRNAs). MicroRNAs (miRNAs) are abundant class of small ncRNAs with 21-25 nucleotides length. The first miRNA was discovered in 1993, and miRNA research was further accelerated by the Fire and Mello’s discovery of RNA interference (RNAi) in 1998.
In mammalian cells, the biogenesis of miRNAs is mediated by multiple biochemical steps including processing of miRNA precursors by two RNase III enzymes, Drosha and Dicer, and formation of RNA-induced silencing complex with Argonaute (Ago) proteins. miRNAs embedded in Ago proteins recognize their binding sequences predominantly located in 3’UTRs of target mRNAs through sequence complementarity between 5’ seed regions of miRNAs (nt 2-7) and their binding sites and suppress their expression. Reflecting the short sequence dependency, individual miRNAs regulate hundreds of target genes, thus forming complex gene regulatory networks. In addition to regulation of normal cells, miRNAs are known to modulate various pathological processes including cancer. We have investigated the relationships between cancer, miRNA biogenesis, and miRNA-mediated gene regulation and revealed the roles of tumor suppressor p53 in miRNA processing (Suzuki et al, Nature, 2009; Suzuki et al, Mol Cell, 2011).
In cancer biology, numerous studies have reported that miRNAs regulate many aspects of autonomous behavior of cancer cells, including sustained proliferation, resistance to cell death, and acquisition of metastasis phenotypes. We also previously reported that miRNAs regulate immune phenotypes of malignant lymphoma cells and modulate surrounding tumor microenvironments, illuminating miRNA-mediated non-cell-autonomous mechanism in cancer progression (Matsuyama et al, Blood, 2011; Suzuki et al, Oncogene, 2015).
6.Systems RNA Biology: Can we describe RNA and genome functions using mathematical equations and computers?
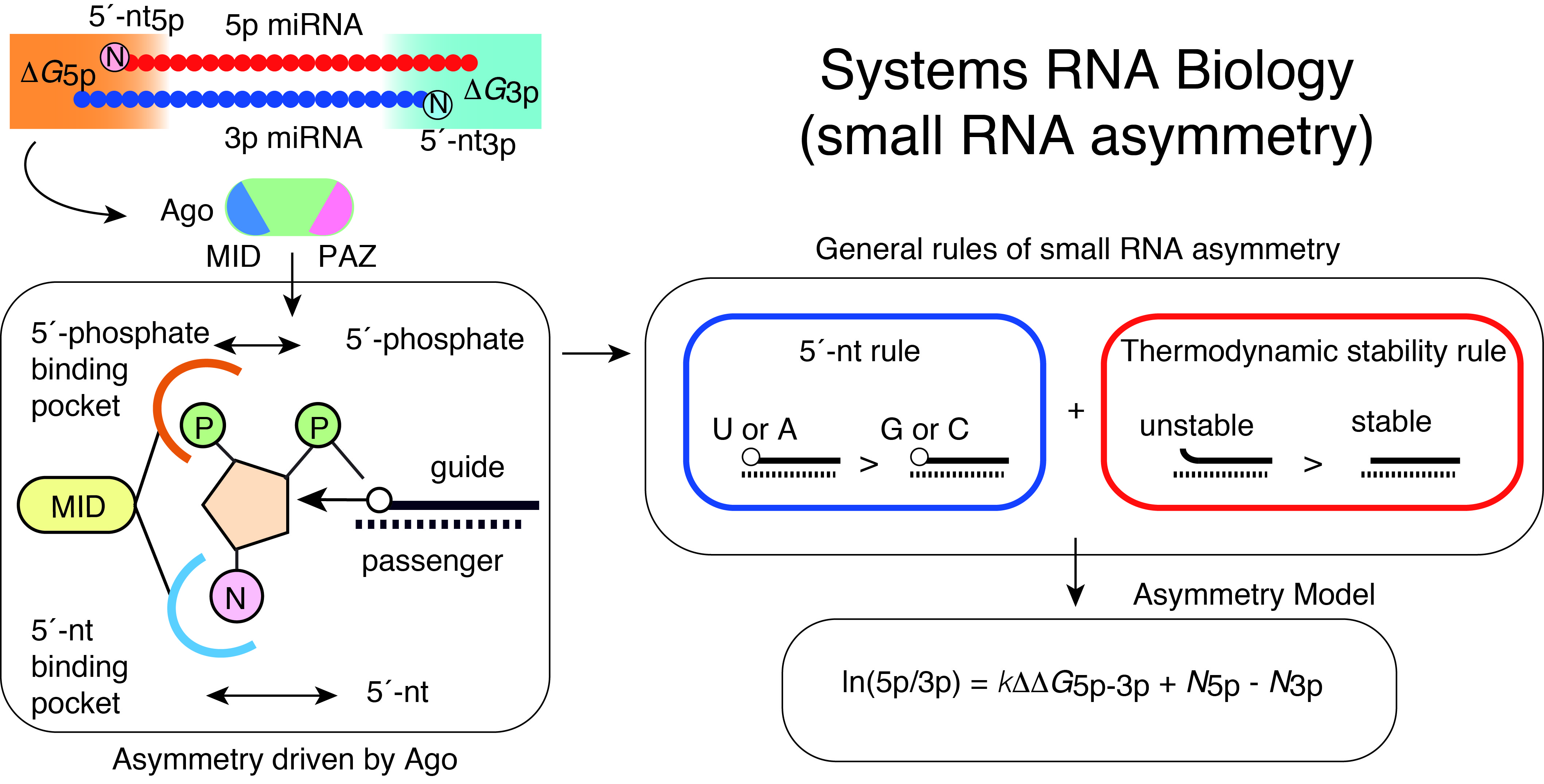
Gene regulation by miRNAs is complicated with targeting of multiple mRNAs by individual miRNAs and their generally mild effects on target mRNAs. Understanding of such complex systems requires sophisticated approaches integrating multiple genome-wide datasets including transcriptomes. We have previously developed analytical approaches to infer miRNA activities from mRNA expression datasets and demonstrated that applying these approaches to analysis of paired miRNA-mRNA datasets improves identification of robust bio-markers in cancer (Suzuki et al, Nucleic Acid Res, 2013; Suzuki et al, Leukemia, 2013).
Of the two strands of miRNA duplexes, one miRNA strand is incorporated into an Ago protein, while the other strand is degraded. Asymmetric selection of one guide strand is crucial in miRNA-mediated gene regulation and specificity of RNAi. By combining molecular biology, biophysics, structural biology, and bioinformatics, we have presented a molecular and theoretical framework that describes the general features and underlying mechanisms of miRNA asymmetry and further presents modeling to estimate the asymmetry (Suzuki et al, Nat Struct Mol Biol, 2015; Matsuyama et al, IJMS, 2019). In consistent with the prediction by this model, cancer-associated miRNA variations were shown to modulate asymmetric selection. This model has contributed to development of synthetic RNA-Based immunomodulatory gene circuits for cancer immunotherapy (Nissim et al, Cell, 2017) and discovery of the first case of a pathogenic gain-of-function miRNA mutation in human diseases (Grigelioniene et al, Nature Medicine, 2019).
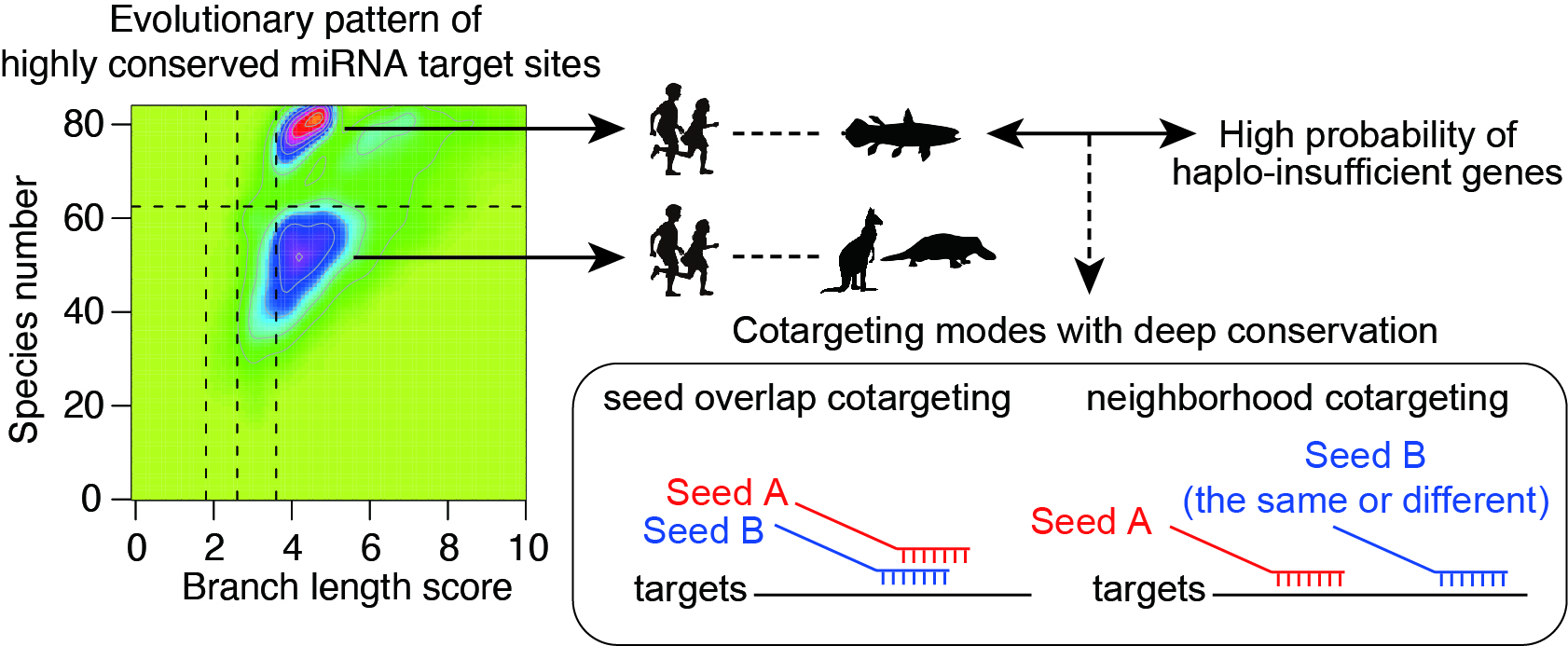
Combinatorial gene regulation by multiple miRNAs is widespread and closely spaced target sites often act cooperatively to achieve stronger repression (“neighborhood” miRNA cotargeting). Based on the functional studies of two miRNA alterations in disease pathogenesis, we have shown that miRNA cotargeting using extensively overlapping target sites (“seed overlap” miRNA cotargeting) increases susceptibility to downregulation by two miRNAs, consistent with additive but not cooperative recruitment of two miRNAs. Systematic characterization further revealed that extensive “seed overlap” is a prevalent feature among broadly conserved miRNAs. Intriguingly, highly conserved target sites of broadly conserved miRNAs are largely divided into two classes—those conserved among eutherian mammals and from human to Coelacanth, and the latter has a stronger association with both “seed overlap” and “neighborhood” miRNA cotargeting. These findings collectively suggest the complexity of distinct modes of miRNA cotargeting and the importance of their perturbations in human diseases (Kitai et al, BMC Biology, 2022).
7. beyond
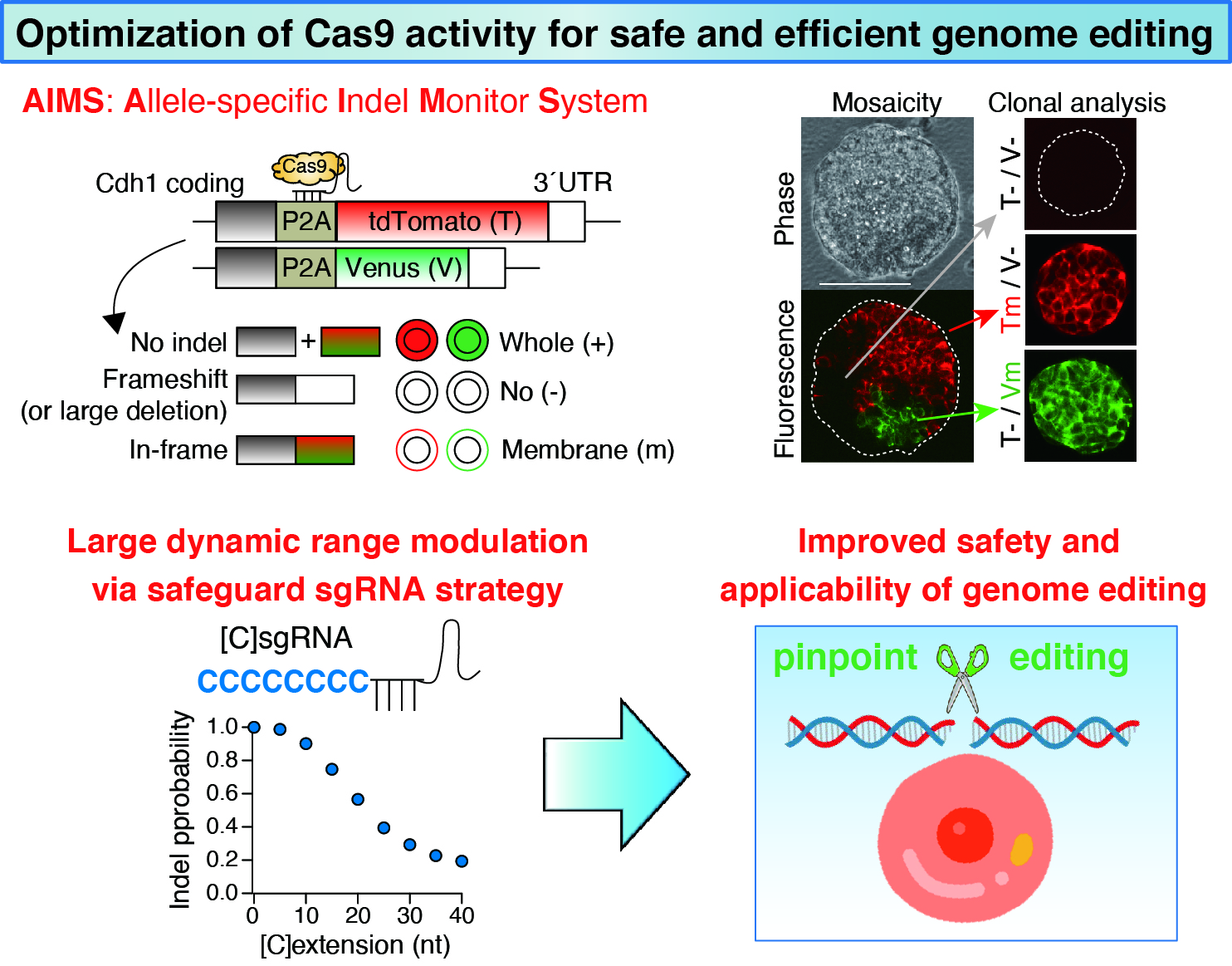
By expanding the expertise in computational modeling in previous studies, a recent collaboration project with Kyushu University has successfully developed new CRISPR-based genome editing strategy, called “safeguard sgRNA” strategy, which improves the safety and applicability of genome editing (Kawamata et al, Nature Biomedical Engineering, 2023).